

Fasta文件生成详解

Fasta文件生成详解:Fasta是一种常用的生物信息学文件格式,用于存储核酸或蛋白质序列数据。其生成过程包括序列提取、格式转换和文件保存等步骤。从原始数据中提取出所需的序列信息;将提取的序列信息转换成Fasta格式;将转换后的数据保存为Fasta文件。在生成Fasta文件时,需要注意序列的准确性和完整性,以及文件格式的规范性,以确保后续生物信息学分析的顺利进行。
在生物信息学和分子生物学领域,Fasta文件是一种常见的序列数据格式,它被广泛应用于基因组学、转录组学、蛋白质组学等研究领域,用于存储和分享大量的核酸或蛋白质序列信息,本文将详细介绍Fasta文件的生成方法,帮助读者了解如何创建和利用这种重要的数据格式。
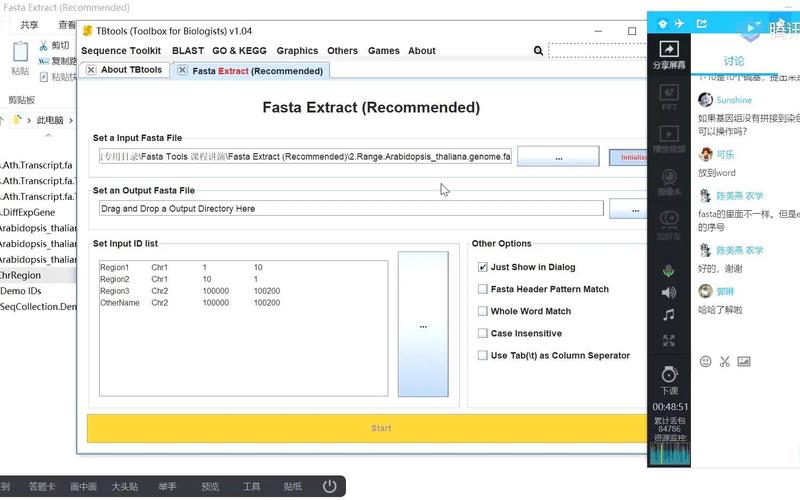
Fasta文件概述
Fasta文件是一种文本文件格式,用于存储生物序列数据,每个Fasta文件包含一个或多个序列,每个序列以“>”符号开始,后面跟着序列的描述信息(可选)和序列本身,描述信息可以是关于序列的任何注释或说明,而序列本身则是由字母(通常是A、C、G、T或其变体)组成的字符串。
Fasta文件生成步骤
1、收集序列数据:你需要收集要存储在Fasta文件中的序列数据,这些数据可以是来自公共数据库的序列,也可以是实验测序得到的数据。
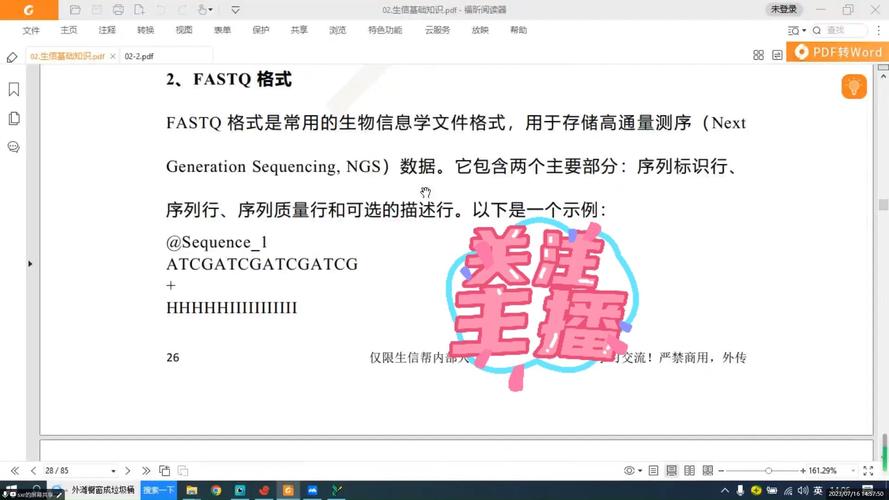
2、准备描述信息:为每个序列准备描述信息,描述信息可以是关于序列的来源、功能或其他相关信息,这些信息将出现在Fasta文件的头部,以“>”符号开始。
3、使用文本编辑器创建Fasta文件:打开一个文本编辑器(如Notepad、TextEdit或任何其他你喜欢的文本编辑器),创建一个新文件。

4、输入序列和描述信息:在文本编辑器中,按照Fasta文件的格式输入序列和描述信息,每个序列应以“>”符号开始,后面跟着描述信息,然后是序列本身,多个序列可以按此格式连续输入。
5、保存文件:在文本编辑器中,将文件保存为Fasta格式,Fasta文件的扩展名为“.fasta”或“.fa”,确保在保存时选择文本或纯文本格式,以避免包含任何不必要的字符或格式。
使用生物信息学软件生成Fasta文件
除了使用文本编辑器手动创建Fasta文件外,还可以使用生物信息学软件来生成Fasta文件,这些软件通常具有从其他格式(如FASTQ、SAM等)转换或导出为Fasta格式的功能,以下是使用生物信息学软件生成Fasta文件的一般步骤:
1、选择合适的软件:根据你的需求和偏好,选择一个适合你的生物信息学软件,一些常用的生物信息学软件包括BLAST、SAMtools等。
2、打开软件并导入数据:打开所选软件,并导入你要转换的数据,这通常可以通过文件菜单中的“打开”或“导入”选项来完成。
3、选择导出为Fasta格式:在软件中找到导出或转换选项,并选择Fasta格式作为输出格式,确保选择正确的参数和选项,以便导出正确的数据。
4、保存文件:完成导出后,软件将生成一个Fasta文件,你可以选择保存该文件到你的计算机上,以便后续使用和分析。
注意事项
1、确保数据的准确性:在生成Fasta文件之前,确保你的序列数据是准确的,任何错误或不一致的数据都可能影响后续的分析和解释。
2、使用适当的描述信息:为每个序列提供适当的描述信息,以便于后续分析和理解,描述信息应清晰、准确,并包含有关序列的来源、功能或其他相关信息。
3、遵循Fasta文件格式规范:在创建或转换Fasta文件时,确保遵循Fasta文件的格式规范,这包括使用正确的语法和标记,以及避免包含任何不必要的字符或格式。
4、备份数据:在生成Fasta文件之前,务必备份你的原始数据和中间结果,这有助于防止数据丢失或损坏,并确保你可以随时恢复工作进度。
本文详细介绍了如何生成Fasta文件,包括手动使用文本编辑器创建Fasta文件和使用生物信息学软件生成Fasta文件的步骤,Fasta文件在生物信息学和分子生物学领域具有广泛的应用价值,了解如何生成Fasta文件将有助于更好地进行序列数据分析和研究。



